La Sindrome di Lowe
🥼 Conosci la Sindrome di Lowe? Scopriamo insieme di cosa si tratta!

La sindrome di Lowe (OMIM #309000) è una rara malattia multi-sistemica con prevalenza molto bassa, stimata tra 1: 500.000 e 1:1.000.000 secondo i dati dell’American Lowe's Syndrome Association e dell’Associazione italiana della sindrome di Lowe; descritta per la prima volta nel 1952 da Charles Lowe ed i suoi colleghi, grazie a studi condotti su tre pazienti di età compresa tra 2-7 mesi e seguiti fino a 1-4 anni.
Caratteristiche generali
Inizialmente, la combinazione delle caratteristiche cliniche dei pazienti di tali studi non rispecchiava nessuna delle patologie fino ad allora descritte. Però, grazie ai successivi studi condotti da Richards e la sua equipe nel 1965 si è giunti alla conclusione che vi fosse un modello di ereditarietà dovuto ad una mutazione recessiva legata al cromosoma X. Infatti, Nel 1992 viene identificato sul cromosoma X il gene coinvolto nelle manifestazioni fenotipiche caratteristiche della malattia, ovvero OCRL1 (Xq26.1).
Il gene OCRL1 codifica per una delle 10 inositolo 5-fosfatasi presenti nei vertebrati, nota come OCRL, i cui substrati sono rappresentati dai fosfoinositidi. Specificamente, i fosfoinositidi sono fosfolipidi di membrana implicati nella regolazione di diversi processi cellulari, ed in natura ne sono stati descritte 7 tipologie differenti generate dalla fosforilazione reversibile del proprio anello di inositolo in posizione 3’, 4’ e 5’. In particolare, la fosfatasi OCRL è coinvolta nella rimozione del gruppo fosfato in posizione 5’ dell’anello di inositolo dei suoi substrati, in particolare del fosfoinositide PI(4,5)P2 (fosfatidilinositolo-4,5‑bisfosfato).
Nella sindrome di Lowe, le mutazioni nella sequenza genica di OCRL1 aboliscono o riducono l’attività di OCRL comportano un aumento dei livelli intracellulari di PI(4,5)P2 che si traduce in alterazioni di vari processi cellulari, comprendenti difetti nel traffico intracellulare conseguenti alla errata composizione lipidica delle membrane, ridotta divisione cellulare e alterata architettura del citoscheletro di actina.
Genetica
Grazie ai diversi studi condotti negli anni possiamo affermare, quindi, che la sindrome di Lowe è una malattia a trasmissione X-linked recessiva causata da mutazioni che interessano il gene OCRL1, codificante per l’inositolo 5-fosfatasi OCRL, presente nella regione cromosomica Xq26.1.
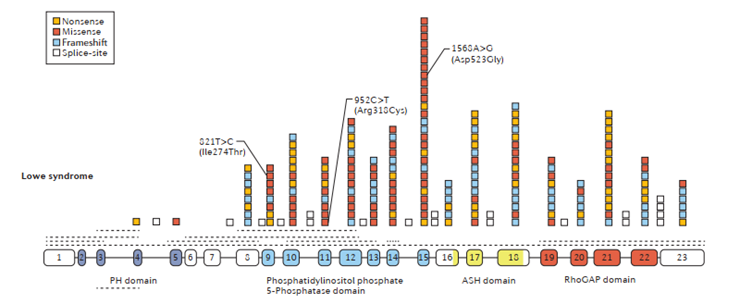
OCRL1 è costituito a livello genomico da 23 esoni e 22 introni e nel corso del tempo sono state identificate più di 170 mutazioni differenti che si distribuiscono lungo tutta la sequenza del gene. In particolare, le mutazioni descritte, si concentrano agli esoni codificanti il dominio catalitico della proteina OCRL e le regioni al 3’ dello stesso, implicate nelle interazioni della fosfatasi con i diversi interattori. Quindi, questo si traduce nella perdita o ridotta funzione della proteina codificata da OCRL1.
Inoltre, essendo il gene causativo della malattia localizzato sul cromosoma X, tutti gli uomini con mutazioni in OCRL1 mostrano le caratteristiche fenotipiche della sindrome di Lowe, mentre le donne possono essere portatrici (94% dei casi) con la tessa probabilità del 25% di avere un figlio maschio affetto, un figlio maschio sano, una figlia non portatrice o una figlia portatrice.
Il locus genetico è stato identificato grazie alle analisi condotte su due donne con fenotipo caratteristico della sindrome di Lowe e nelle quali è stata rilevata una traslocazione bilanciata dell'autosoma X che coinvolgeva la regione Xq26.
Caratteristiche cliniche
La sindrome di Lowe presenta uno spettro clinico vasto e gravità del fenotipo variabile tra pazienti. Tra le altre manifestazioni cliniche associate alla sindrome abbiamo un ipotonia muscolare generalizzata, ritardo della crescita, artropatia, criptorchidismo, disfunzione piastrinica e anomalie comportamentali.
Inoltre, questa patologia è anche nota come sindrome oculo-cerebro-renale per il principale coinvolgimento di occhi, sistema nervoso centrale e reni in cui si verificano rispettivamente cataratta congenita, disabilità intellettiva e sindrome renale di Fanconi incompleta (disfunzione de tubulo contorto prossimale con alterato riassorbimento di ioni e proteine a basso peso molecolare).
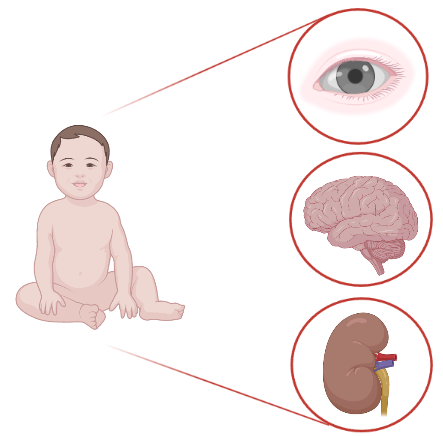
Occhi - cataratta congenita e glaucoma: La maggior parte dei pazienti presenta cataratta bilaterale alla nascita. Infatti, i difetti nelle cellule del cristallino compaiono all'inizio dell'embriogenesi e sono verosimilmente causati da alterazioni nei processi di formazione delle fibre del cristallino primario, che possono portare ad una mancata formazione delle stesse oppure ad una loro rapida degenerazione. Mentre nei pazienti di sesso maschile sono interessate tutte le cellule del cristallino, nelle femmine portatrici si verifica un aumento dell’opacità del cristallino legata all'inattivazione casuale di uno dei due cromosomi X. Inoltre, circa la metà dei pazienti sviluppa glaucoma entro il primo anno.
Sistema nervoso centrale - disabilità intellettiva: i pazienti Lowe hanno disabilità intellettive con entità variabile da lieve a molto grave, e un quoziente intellettivo (QI) pari o inferiore a 50. Infatti, nel 10-25% dei casi si registra una disabilità intellettiva normale o borderline, nel 25% la gravità del ritardo va da lieve a moderata e nel 50-65% si verifica una disabilità intellettiva da grave a profonda. Inoltre, durante l'adolescenza la maggior parte dei soggetti affetti manifesta un comportamento aggressivo, irritabilità, scoppi di rabbia e comportamenti ossessivo-compulsivi.
Rene - Sindrome renale di Fanconi: il fenotipo renale inizia a manifestarsi nei primi mesi di vita dei pazienti ed è caratterizzato da un progressivo peggioramento fino all’insorgenza dei segni caratteristici di una sindrome renale di Fanconi incompleta. Infatti, in tutti i soggetti affetti si verificano proteinuria a basso peso molecolare e albuminuria, bicarbonaturia e acidosi tubulare renale, fosfaturia con ipofosfatemia e rachitismo, perdita di sodio e potassio e poliuria per un'eccessiva perdita di soluti nelle urine. Molto raramente si osserva una perdita renale di glucosio, sintomo invece associato alla sindrome di Fanconi renale con spettro completo.
Gestione e trattamento dei sintomi
Per il trattamento della sindrome di Lowe ci si avvale di un approccio multidisciplinare a seconda della gravità dei sintomi caratteristici presentati da ogni paziente, questo consentirà di intervenire in modo mirato ad ogni alterazione attraverso l’uso di farmaci, intervento chirurgico e/o altre misure. Ad esempio, nel caso del trattamento della cataratta è importante intervenire entro le prime settimane di vita per la rimozione chirurgica, dopo la quale i pazienti potranno utilizzare occhiali da vista o lenti a contatto.
Per la gestione dell’ipotonia e delle sue possibili complicanze è importante iniziare il prima possibile delle terapie riabilitative. Invece, per tenere sotto controllo i comportamenti aggressivi ed ossessivo-compulsivi e per favorire l’apprendimento vi sono programmi pedagogici e psicologici per i ragazzi affetti.
Infine, per quanto riguarda le complicanze renali, solitamente si usano supplementi somministrati oralmente (ad esempio supplementi di fosfato e 1,25-idrossivitamina-D3), negli adulti si procede con la dialisi e solo in rari casi si può effettuare un trapianto di rene.
Lidia Caserta
Fonti:
· Lowe, C. U., Terrey, M., M. & Mac, L. E. (1952). Organic-aciduria, decreased renal ammonia production, hydrophthalmos, and mental retardation; a clinical entity. AMA Am J Dis Child, 83(2),164-84. https://doi.org/10.1001/archpedi.1952.02040060030004;
· Richards, W., Donnell, G. N., Wilson, W. A., Stowens, D., Perry, T. (1965). The Oculo-Cerebro-Renal Syndrome of Lowe. Am J Dis Child, 109(3), 185–203. Doi:10.1001/archpedi.1965.02090020187001;
· De Matteis, M. A., Staiano, L., Emma, F., & Devuyst, O. (2017). The 5-phosphatase OCRL in Lowe syndrome and Dent disease 2. Nature Reviews Nephrology, 13(8), 455–470. https://doi.org/10.1038/nrneph.2017.83;
· Kruger, S. J., Wilson, M. D., Hutchinson, A. K., Peterseim, M. M., Bartholomew, L. R., Saunders, R. A. (2003). Cataracts and Glaucoma in Patients With Oculocerebrorenal Syndrome. Archives of Ophthalmology, 121(9), 1234. https://doi.org/10.1001/archopht.121.9.1234;
· Loi, M. (2006). Lowe syndrome. Orphanet Journal of Rare Diseases, 1(1), 16. https://doi.org/10.1186/1750-1172-1-16;
· Kenworthy, L., Park, T., & Charnas, L. R. (1993). Cognitive and behavioral profile of the oculocerebrorenal syndrome of Lowe. American Journal of Medical Genetics, 46(3), 297–303. https://doi.org/10.1002/ajmg.1320460312.
Ti è piaciuto l'articolo?
 BioDaily.it non riceve alcun contributo pubblico né ospita alcuna pubblicità, quindi si sostiene esclusivamente grazie alle donazioni dei lettori. Ti ringraziamo qualora tu volessi fare una donazione al nostro progetto, puoi farlo cliccando su questo messaggio.
BioDaily.it non riceve alcun contributo pubblico né ospita alcuna pubblicità, quindi si sostiene esclusivamente grazie alle donazioni dei lettori. Ti ringraziamo qualora tu volessi fare una donazione al nostro progetto, puoi farlo cliccando su questo messaggio.